Lataa esitys
Esittely latautuu. Ole hyvä ja odota

1
Sydänpotilaiden hoito, jälkihoito ja sekundaaripreventio
Pohjanmaan Rannikkoseudun Laatuverkoston koulutustilaisuus Hotelli Silveria

3
Kuolinsyyt Suomessa v. 2001

4
Kuolleisuus sydän- ja verisuonitauteihin Suomessa/100 000 asukasta

5
Kuolleisuus sydän- ja verisuonitauteihin Suomessa v. 1999 ikäryhmittäin

6
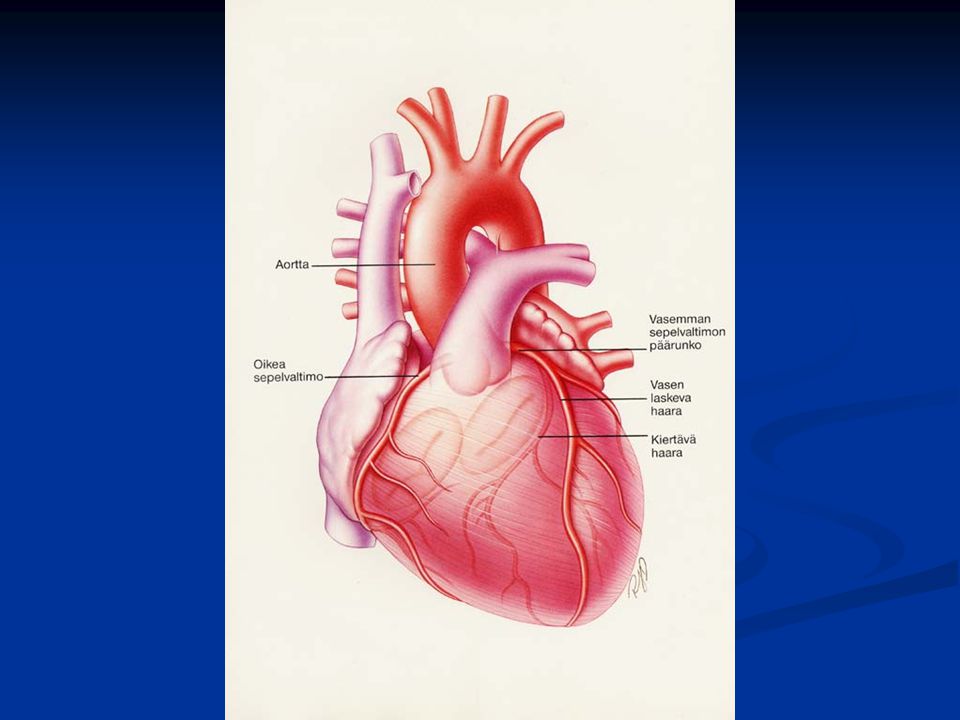
Sepelvaltimotaudin ilmenemismuotoja
Stabiilioireinen sepelvaltimotauti Epästabiilioireinen sepelvaltimotauti Sydäninfarkti Ilman ST-nousua ekg:ssa ST-nousuinfarkti Ilmenemismuoto riippuu sepelvaltimoahtauman luonteesta Sepelvaltimoahtauman luonne vaikuttaa hoidon valintaan

7
Normaali valtimoseinämä
Slide 1: The normal artery wall Endoteelisolut The wall of a normal, healthy artery consists of three distinct layers1. The intima is the innermost layer. This comprises a monolayer of endothelial cells supported on a basement membrane and sub-endothelial matrix. Unlike that of other mammals, the human intima also contains a small number of vascular smooth muscle cells (VSMCs)2. The intermediate layer of the artery wall – the media – consists of concentrically arranged VSMCs in an extracellular matrix of collagen, elastic fibres and glycosaminoglycans1. The outermost structural layer of the artery wall is the adventitia. This comprises an extracellular matrix of longitudinally arranged collagen fibres and contains the vasa vasorum1. Endothelial cells synthesize and secrete a number of substances that control vascular tone. These include the vasodilators nitric oxide (NO), prostacyclin and the prostaglandins PGE2 and PGD2, as well as vasoconstrictors such as endothelin-1, thromboxane A2 and the prostaglandin endoperoxides PGG2 and PGH21,3. The endothelium also expresses substances that regulate the adhesion of monocytes and platelets, and control coagulation through a balance of inhibitory and promotional effects4. In the normal, healthy artery wall, the balance is such that antithrombotic, anti-inflammatory and vasodilatory characteristics predominate1,4. The endothelium is made up of three distinct layers: the intima, the media and the adventitia Endothelial cells secrete substances that control vascular tone Under basal conditions, the endothelium presents an antithrombotic surface, and vasodilatory tendencies predominate References 1 Vallance P. Vascular endothelium, its physiology and pathophysiology. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn, Oxford Medical Publications, Oxford, UK, 1996; 2: 2295–2300. 2 Schwartz SM, DeBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res 1995; 77: –465. 3 Celemajer DS. Endothelial dysfunction: does it matter? Is it reversible? J Am Coll Cardiol 1997; 30: 325–333. 4 Verrier ED, Boyle EM. Endothelial cell injury in cardiovascular surgery: an overview. Ann Thorac Surg 1996; 64: S2–S8. Sileät lihassolut
2. The intermediate layer of the artery wall – the media – consists of concentrically arranged VSMCs in an extracellular matrix of collagen, elastic fibres and glycosaminoglycans1. The outermost structural layer of the artery wall is the adventitia. This comprises an extracellular matrix of longitudinally arranged collagen fibres and contains the vasa vasorum1. Endothelial cells synthesize and secrete a number of substances that control vascular tone. These include the vasodilators nitric oxide (NO), prostacyclin and the prostaglandins PGE2 and PGD2, as well as vasoconstrictors such as endothelin-1, thromboxane A2 and the prostaglandin endoperoxides PGG2 and PGH21,3. The endothelium also expresses substances that regulate the adhesion of monocytes and platelets, and control coagulation through a balance of inhibitory and promotional effects4. In the normal, healthy artery wall, the balance is such that antithrombotic, anti-inflammatory and vasodilatory characteristics predominate1,4. The endothelium is made up of three distinct layers: the intima, the media and the adventitia. Endothelial cells secrete substances that control vascular tone. Under basal conditions, the endothelium presents an antithrombotic surface, and vasodilatory tendencies predominate. References. 1 Vallance P. Vascular endothelium, its physiology and pathophysiology. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn, Oxford Medical Publications, Oxford, UK, 1996; 2: 2295– Schwartz SM, DeBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res 1995; 77: 445– Celemajer DS. Endothelial dysfunction: does it matter Is it reversible J Am Coll Cardiol 1997; 30: 325– Verrier ED, Boyle EM. Endothelial cell injury in cardiovascular surgery: an overview. Ann Thorac Surg 1996; 64: S2–S8. Sileät lihassolut.")
8
Varhainen ateroskleroosi
Slide 2: Early atherosclerosis (I) – Endothelial dysfunction Varhainen ateroskleroosi Lipidi kertyy intimatilaan Endoteelisolujen toimintahäiriö Endothelial dysfunction is believed to be a key element in atherogenesis, occurring in the early stages of the disease process1,2. The conventional risk factors for coronary artery disease, such as smoking, hypertension, diabetes and hyperlipidaemia, have also been found to be associated with endothelial dysfunction. Particular attention has been focused on the role played by elevated cholesterol levels in endothelial function, and significant correlations have been reported between plasma levels of low-density lipoprotein (LDL) and the degree of endothelial impairment1. It is believed that LDL usually enters the intima by means of transcytosis, and is subsequently oxidized by oxidative free radicals to form oxidized LDL (oxLDL)2. The accumulation of oxLDL within the intima is associated with abnormalities in a number of endothelial functions, including impairment of the endothelial-dependent vasodilatory response to agents such as acetylcholine, histamine and serotonin3, or to hyperaemic blood flow4. Endothelial dysfunction occurs early in atherogenesis Elevated plasma lipid levels are a known risk factor for atherosclerosis and are directly related to the degree of endothelial impairment Lipid accumulation within the intima is associated with abnormal endothelial cell function and impairment of the endothelial- dependent vasodilatory response to various stimuli References 1 Celermajer DS. Endothelial dysfunction: does it matter? Is it reversible? J Am Coll Cardiol 1997; 30: 325–333. 2 Scott J. Atheroma, the vessel wall, and thrombosis. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2289–2295. 3 Ruschitzka FT, Noll G, Lüscher TF. The endothelium in coronary artery disease. Cardiology 1997; 88 (suppl 3): 3–19. 4 Clarkson P, Celermajer DS, Powe AJ et al. Endothelium-dependent dilatation is impaired in young healthy subjects with a family history of premature coronary disease. Circulation 1997; 96: 3378–3383. Lipid
 – Endothelial dysfunction. Varhainen ateroskleroosi. Lipidi kertyy intimatilaan Endoteelisolujen toimintahäiriö. Endothelial dysfunction is believed to be a key element in atherogenesis, occurring in the early stages of the disease process1,2. The conventional risk factors for coronary artery disease, such as smoking, hypertension, diabetes and hyperlipidaemia, have also been found to be associated with endothelial dysfunction. Particular attention has been focused on the role played by elevated cholesterol levels in endothelial function, and significant correlations have been reported between plasma levels of low-density lipoprotein (LDL) and the degree of endothelial impairment1. It is believed that LDL usually enters the intima by means of transcytosis, and is subsequently oxidized by oxidative free radicals to form oxidized LDL (oxLDL)2. The accumulation of oxLDL within the intima is associated with abnormalities in a number of endothelial functions, including impairment of the endothelial-dependent vasodilatory response to agents such as acetylcholine, histamine and serotonin3, or to hyperaemic blood flow4. Endothelial dysfunction occurs early in atherogenesis. Elevated plasma lipid levels are a known risk factor for atherosclerosis and are directly related to the degree of endothelial impairment. Lipid accumulation within the intima is associated with abnormal endothelial cell function and impairment of the endothelial- dependent vasodilatory response to various stimuli. References. 1 Celermajer DS. Endothelial dysfunction: does it matter Is it reversible J Am Coll Cardiol 1997; 30: 325– Scott J. Atheroma, the vessel wall, and thrombosis. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2289– Ruschitzka FT, Noll G, Lüscher TF. The endothelium in coronary artery disease. Cardiology 1997; 88 (suppl 3): 3–19. 4 Clarkson P, Celermajer DS, Powe AJ et al. Endothelium-dependent dilatation is impaired in young healthy subjects with a family history of premature coronary disease. Circulation 1997; 96: 3378–3383. Lipid.")
9
Varhainen ateroskleroosi – endoteelidysfunktio
Slide 3: Early atherosclerosis (II) – The consequences of endothelial dysfunction Varhainen ateroskleroosi – endoteelidysfunktio Endoteelisolujen aktivaatio -adheesiomolekyylit -tulehdussolujen (monosyytit) kertyminen In addition to alterations in vasomotor control, endothelial dysfunction leads to the up-regulation of leukocyte and endothelium adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), resulting in increased adherence of inflammatory cells (principally monocytes and T-lymphocytes). Further effects of endothelial dysfunction include increased permeability of the endothelial layer to monocytes/macrophages and lipoproteins, and increased migration and proliferation of VSMCs1,2. Endothelial cell dysfunction leads to the up-regulation of adhesion molecules and increased permeability of the endothelium to inflammatory cells and lipoproteins References 1 Celermajer DS. Endothelial dysfunction: does it matter? Is it reversible? J Am Coll Cardiol 1997; 30: 325–333. 2 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol EJ. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441–460. Lipid
 – The consequences of endothelial dysfunction. Varhainen ateroskleroosi – endoteelidysfunktio. Endoteelisolujen aktivaatio. -adheesiomolekyylit. -tulehdussolujen (monosyytit) kertyminen. In addition to alterations in vasomotor control, endothelial dysfunction leads to the up-regulation of leukocyte and endothelium adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), resulting in increased adherence of inflammatory cells (principally monocytes and T-lymphocytes). Further effects of endothelial dysfunction include increased permeability of the endothelial layer to monocytes/macrophages and lipoproteins, and increased migration and proliferation of VSMCs1,2. Endothelial cell dysfunction leads to the up-regulation of adhesion molecules and increased permeability of the endothelium to inflammatory cells and lipoproteins. References. 1 Celermajer DS. Endothelial dysfunction: does it matter Is it reversible J Am Coll Cardiol 1997; 30: 325– Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol EJ. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441–460. Lipid.")
10
Ateroskleroottisen plakin muodostuminen valtimon seinämään
Slide 6: Pathology of atherosclerosis (I) – Formation of the early atherosclerotic lesion Monosyytit aktivoituvat makrofaageiksi joista kehittyy vaahtosoluja Endoteeliin kertyneet monosyytit Makrofaagit houkuttelevat sileitä lihassoluja intimaan Lipid core The events discussed thus far provide a brief overview of the complex processes involved in the development of the early atherosclerotic lesion, which represents the first histological manifestation of atherosclerotic disease1. The key steps in its pathogenesis can be summarized as follows: Entrance of lipid into the intimal space2, where it is oxidized by free radical species3 Impairment of endothelial cell function, following chronic injury by agents such as oxLDL or nicotine3 Recruitment of monocytes and their subsequent activation to macrophages within the intima4 Engulfment of oxLDL by intimal macrophages, resulting in the formation of cholesterol-rich foam cells2 VSMC recruitment, migration into the intima and proliferation, facilitating the increased production of extracellular matrix materials1. Endothelial dysfunction is associated with an increased permeability to lipids and inflammatory cells Monocytes recruited into the intima are activated to macrophages, which ingest oxLDL to form cholesterol-rich foam cells The recruitment and proliferation of VSMCs (mediated by growth factors) enables the production of extracellular materials that are incorporated in the advanced atherosclerotic plaque References 1 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 2 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441–460. 3 Scott J. Atheroma, the vessel wall, and thrombosis. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2289–2295. 4 Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. Adventitia
 – Formation of the early atherosclerotic lesion. Monosyytit aktivoituvat makrofaageiksi joista kehittyy vaahtosoluja. Endoteeliin kertyneet monosyytit. Makrofaagit houkuttelevat sileitä lihassoluja intimaan. Lipid core. The events discussed thus far provide a brief overview of the complex processes involved in the development of the early atherosclerotic lesion, which represents the first histological manifestation of atherosclerotic disease1. The key steps in its pathogenesis can be summarized as follows: Entrance of lipid into the intimal space2, where it is oxidized by free radical species3. Impairment of endothelial cell function, following chronic injury by agents such as oxLDL or nicotine3. Recruitment of monocytes and their subsequent activation to macrophages within the intima4. Engulfment of oxLDL by intimal macrophages, resulting in the formation of cholesterol-rich foam cells2. VSMC recruitment, migration into the intima and proliferation, facilitating the increased production of extracellular matrix materials1. Endothelial dysfunction is associated with an increased permeability to lipids and inflammatory cells. Monocytes recruited into the intima are activated to macrophages, which ingest oxLDL to form cholesterol-rich foam cells. The recruitment and proliferation of VSMCs (mediated by growth factors) enables the production of extracellular materials that are incorporated in the advanced atherosclerotic plaque. References. 1 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 2 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441– Scott J. Atheroma, the vessel wall, and thrombosis. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2289– Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. Adventitia.")
11
Sidekudoskannen kehittyminen
Slide 7: The fibrous cap of the developing atheromatous plaque Lihassolut muuttuvat supistuvista “korjaaviksi” ja erittävät soluväliainetta plakin kanteen Lipid core The most obvious difference between early and more developed atherosclerotic lesions is the incorporation into the latter of a fibrous cap. This cap covers the lipid-rich inner core and separates it from blood circulating through the lumen. The fibrous cap that characterizes the more advanced atheromatous lesion comprises a dense extracellular matrix generated by repair phenotype VSMCs1–3. Indeed, VSMCs are the only cells within an atherosclerotic plaque that are capable of synthesizing and maintaining the fibrous cap4. The fibrous cap is usually by far the largest component of the plaque, occupying more than 70% of the total volume of a typical stenotic coronary lesion5,6. The principal components are interstitial fibrillar collagen, elastin, proteoglycans and glycosaminoglycans3,5,7. Advanced plaques include a fibrous cap composed of extracellular matrix materials The fibrous cap separates the lipid-rich plaque core from the circulating blood, stabilizing the lesion VSMCs are the only cells capable of synthesizing the fibrous cap that stabilizes the atherosclerotic plaque References 1 Scott J. Atheroma, the vessel wall, and thrombosis. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2289–2295. 2 Shanahan CM, Weissberg PL. Smooth muscle cell heterogeneity: patterns of gene expression in vascular smooth muscle cells in vitro and in vivo. Arterioscler Thromb Vasc Biol 1998; 18 (3): 333–338. 3 Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844–2850. 4 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 5 Shah PK. New insights into the pathogenesis and prevention of acute coronary syndromes. Am J Cardiol 1997; 79: 17–23. 6 Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657–671. 7 Crea F, Biasucci LM, Buffon A. Role of inflammation in the pathogenesis of unstable coronary artery disease. Am J Cardiol 1997; 80: 10E–16E. Adventitia Weissberg, 1999
: 333– Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844– Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 5 Shah PK. New insights into the pathogenesis and prevention of acute coronary syndromes. Am J Cardiol 1997; 79: 17–23. 6 Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657– Crea F, Biasucci LM, Buffon A. Role of inflammation in the pathogenesis of unstable coronary artery disease. Am J Cardiol 1997; 80: 10E–16E. Adventitia. Weissberg,")
12
Slide 10: Characteristics of the stable atherosclerotic plaque
Stabiili plakki Slide 10: Characteristics of the stable atherosclerotic plaque Kansi Lihassolut Endoteeli Lipid core An atherosclerotic plaque is considered to be stable if the risk of rupture, with all its consequences, is low. Studies have identified a number of features that are common to stable plaques. These include: A thick fibrous cap of uniform density that is able to confer mechanical stability on the overall structure by reducing circumferential tensile stress1,2 A high VSMC and collagen content in the fibrous cap2,3 A lipid-rich core that occupies less than 40% of the total volume of the plaque4 A relatively low infiltration of inflammatory cells (principally macrophages and T-lymphocytes)2–5. A stable atherosclerotic plaque is at low risk of rupture Characteristic features of a stable plaque include a thick fibrous cap with a high VSMC and collagen content, a lipid-rich core that occupies a relatively small volume, and a low inflammatory cell content References 1 Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999; 340: 115–126. 2 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 3 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402–417. 4 Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. 5 Kinlay S, Ganz P. Role of endothelial dysfunction in coronary artery disease and implications for therapy. Am J Cardiol 1997; 80: 11I–16I. Adventitia Median lihassolut (supistuva tyyppi)
2–5. A stable atherosclerotic plaque is at low risk of rupture. Characteristic features of a stable plaque include a thick fibrous cap with a high VSMC and collagen content, a lipid-rich core that occupies a relatively small volume, and a low inflammatory cell content. References. 1 Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999; 340: 115– Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 3 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402– Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. 5 Kinlay S, Ganz P. Role of endothelial dysfunction in coronary artery disease and implications for therapy. Am J Cardiol 1997; 80: 11I–16I. Adventitia. Median lihassolut. (supistuva tyyppi)")
13
Stabiili plakki Paksu, runsaasti lihassoluja sisältävä kansi
Slide 11: The stable atherosclerotic plaque Stabiili plakki This slide depicts a stable atherosclerotic plaque with a thick fibrous cap. Note the uniformity of the cap’s density, with no notable thinning at its ends (the ‘shoulders’ of the plaque). It is at these shoulder regions of the plaque that shear stresses are known to be greatest and rupture most commonly occurs1–3. Plaques of this nature may grow to a considerable size without necessarily causing significant narrowing of the arterial lumen. Thus, in spite of their size, it is possible for large atherosclerotic plaques to be asymptomatic and undetectable by angiography. This is facilitated by vascular remodelling, whereby the affected artery increases its external diameter in order to accommodate the growing lesion without notably reducing the dimensions of the lumen4. It is estimated that vascular remodelling can afford an increase in total arterial cross-sectional area of up to 40%5. However, the degree of remodelling that can be achieved varies considerably from patient to patient, and even between arterial segments in the same patient4. Thus, while effective vascular remodelling is a major factor in avoiding the development of arterial stenosis in some patients with atherosclerosis, inadequate compensatory dilatation may contribute to occlusion of the arterial lumen in others. Vascular remodelling can enable significant plaque growth without notable narrowing of the arterial lumen Effective remodelling may be a major factor in the prevention of arterial occlusion in patients with atherosclerosis References 1 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402–417. 2 Kristensen SD, Ravn HB, Falk E. Insights into the pathophysiology of unstable coronary artery disease. Am J Cardiol 1997; 80: 5E–9E. 3 Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657–671. 4 Glasgov S, Weisenberg E, Zarius C et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987; 316: 371–375. 5 Davies MJ, Ho SY. Stable angina. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 107–151. Paksu, runsaasti lihassoluja sisältävä kansi
. It is at these shoulder regions of the plaque that shear stresses are known to be greatest and rupture most commonly occurs1–3. Plaques of this nature may grow to a considerable size without necessarily causing significant narrowing of the arterial lumen. Thus, in spite of their size, it is possible for large atherosclerotic plaques to be asymptomatic and undetectable by angiography. This is facilitated by vascular remodelling, whereby the affected artery increases its external diameter in order to accommodate the growing lesion without notably reducing the dimensions of the lumen4. It is estimated that vascular remodelling can afford an increase in total arterial cross-sectional area of up to 40%5. However, the degree of remodelling that can be achieved varies considerably from patient to patient, and even between arterial segments in the same patient4. Thus, while effective vascular remodelling is a major factor in avoiding the development of arterial stenosis in some patients with atherosclerosis, inadequate compensatory dilatation may contribute to occlusion of the arterial lumen in others. Vascular remodelling can enable significant plaque growth without notable narrowing of the arterial lumen. Effective remodelling may be a major factor in the prevention of arterial occlusion in patients with atherosclerosis. References. 1 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402– Kristensen SD, Ravn HB, Falk E. Insights into the pathophysiology of unstable coronary artery disease. Am J Cardiol 1997; 80: 5E–9E. 3 Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657– Glasgov S, Weisenberg E, Zarius C et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987; 316: 371– Davies MJ, Ho SY. Stable angina. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 107–151. Paksu, runsaasti lihassoluja. sisältävä kansi.")
14
Slide 16: The vulnerable atherosclerotic plaque
“Haavoittuva” plakki Slide 16: The vulnerable atherosclerotic plaque Lipidiydin As discussed in Slide 11, a number of characteristic features are associated with stable atherosclerotic plaques. Plaques that are considered vulnerable and at high risk of rupture are similarly associated with specific characteristics. These include: A lipid core that exceeds 40% of the total volume of the plaque1 A high infiltration of macrophage and T-lymphocyte cells2,3 A thin, friable fibrous cap with a reduced collagen and VSMC content1 Increased circumferential wall stress on the fibrous cap2,3. While vulnerable plaques are believed to account for only 10–20% of all coronary lesions, it has been suggested that they may be responsible for as many as 80–90% of acute clinical events4. Characteristic features of atherosclerotic plaques that are vulnerable to rupture include: a large lipid core, a high infiltration of inflammatory cells, a thin, friable fibrous cap in which the concentrations of collagen and VSMCs are reduced, and increased circumferential wall stress on the fibrous cap Vulnerable plaques account for a comparatively small proportion of all coronary lesions, but are responsible for the vast majority of acute clinical events References 1 Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. 2 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402–417. 3 Lendon CL, Davies MJ, Born GVR, Richardson PD. Atherosclerotic plaque caps are locally weakened when macrophage density is increased. Atherosclerosis 1991; 87: 87–90. 4 Vaughan CJ, Murphy MB, Buckley BM. Statins do more than just lower cholesterol. Lancet 1996; 348: 1079–1082. Adventitia

15
Haavoittuva plakki Kannen lihassolujen jakaantuminen loppuu
Slide 17: Advanced atherosclerosis Haavoittuva plakki Kannen lihassolujen jakaantuminen loppuu Aktivoituneet makrofaagit aiheuttavat lihassolujen kuoleman ja syövyttävät soluväliainetta In advanced atherosclerosis, there is a high risk of rupture of vulnerable plaques. A number of factors contribute to this risk, including the infiltration of large numbers of inflammatory cells – particularly macrophages1,2. Activated macrophages not only structurally weaken the fibrous cap of the plaque but also induce VSMC apoptosis3,4, thus reducing the capacity for plaque repair. Early senescence and slowed proliferation of intimal VSMCs are further features of advanced atherosclerosis that promote plaque instability and rupture, rather than repair1,5. The risk of rupture of vulnerable plaques is high in advanced atherosclerosis Factors such as high levels of macrophage infiltration, and slowed proliferation and enhanced early senescence of VSMCs contribute to this increased risk of plaque disruption References 1 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402–417. 2 Lendon CL, Davies MJ, Born GVR, Richardson PD. Atherosclerotic plaque caps are locally weakened when macrophage density is increased. Atherosclerosis 1991; 87: 87–90. 3 Davies MJ, Ho SY. Atherosclerosis: the process. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 23–61. 4 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 5 Bennett MR, Macdonald K, Chan S et al. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998; 82: 704–712.
: S3–S10. 5 Bennett MR, Macdonald K, Chan S et al. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998; 82: 704–712.")
16
Slide 21: Unstable coronary artery disease (I)
Epästabiili sepelvaltimotauti Verihiutaleet kertyvät repeämäkohtaan Lipid core The term ‘unstable coronary artery disease’ (UCAD) encompasses the coronary syndromes of UA and non-Q-wave myocardial infarction (NQMI)1. Symptoms almost invariably arise from the disruption or erosion of an atherosclerotic plaque, which allows circulating blood to contact the lesion and its highly thrombogenic, lipid-rich core. The resulting sequence of events leads to the adhesion and aggregation of platelets at the site of rupture, and the subsequent formation of a partially occlusive, platelet-rich, ‘white’ thrombus1–3. While plaque disruption and subsequent thrombosis may be symptomatic, giving rise to ischaemia, it has been observed that these events frequently occur silently4. UCAD encompasses the coronary syndromes of UA and NQMI The underlying pathology of UCAD involves atherosclerotic plaque rupture, with the subsequent formation of a platelet- rich, ‘white’ thrombus References 1 Campbell RWF, Wallentin L, Verheugt FWA et al. Management strategies for a better outcome in unstable coronary artery disease. Clin Cardiol 1998; 21: 314–322. 2 Davies MJ. The pathophysiology of ischaemic heart disease. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2318–2321. 3 Giri S, Waters DD. Pathophysiology and initial management of the acute coronary syndromes. Curr Opin Cardiol 1996; 11: 351–360. 4 Davies MJ, Ho SY. The acute ischemic syndromes and coronary disease progression. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 63–106. Reprinted from Weissberg PL. Antianginal medication. In: Schofield PM (ed) Angina Pectoris in Clinical Practice, 1999, with permission from Martin Dunitz Ltd Adventitia Weissberg, 1999
 encompasses the coronary syndromes of UA and non-Q-wave myocardial infarction (NQMI)1. Symptoms almost invariably arise from the disruption or erosion of an atherosclerotic plaque, which allows circulating blood to contact the lesion and its highly thrombogenic, lipid-rich core. The resulting sequence of events leads to the adhesion and aggregation of platelets at the site of rupture, and the subsequent formation of a partially occlusive, platelet-rich, ‘white’ thrombus1–3. While plaque disruption and subsequent thrombosis may be symptomatic, giving rise to ischaemia, it has been observed that these events frequently occur silently4. UCAD encompasses the coronary syndromes of UA and NQMI. The underlying pathology of UCAD involves atherosclerotic plaque rupture, with the subsequent formation of a platelet- rich, ‘white’ thrombus. References. 1 Campbell RWF, Wallentin L, Verheugt FWA et al. Management strategies for a better outcome in unstable coronary artery disease. Clin Cardiol 1998; 21: 314– Davies MJ. The pathophysiology of ischaemic heart disease. In: Weatherall DJ, Ledingham JGG, Warrell DA. Oxford Textbook of Medicine, 3rd Edn. Oxford Medical Publications, Oxford, UK, 1996; 2: 2318– Giri S, Waters DD. Pathophysiology and initial management of the acute coronary syndromes. Curr Opin Cardiol 1996; 11: 351– Davies MJ, Ho SY. The acute ischemic syndromes and coronary disease progression. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 63–106. Reprinted from Weissberg PL. Antianginal medication. In: Schofield PM (ed) Angina Pectoris in Clinical Practice, 1999, with permission from Martin Dunitz Ltd. Adventitia. Weissberg,")
17
Slide 22: Unstable coronary artery disease (II)
Epästabiili sepelvaltimotauti Slide 22: Unstable coronary artery disease (II) Hyytymän muodostuminen Thrombus Lipid core The formation of a platelet-rich, ‘white’ thrombus is the primary physiological response to rupture of an atherosclerotic plaque. If the initial disruption of the plaque is superficial, the thrombus formed will be mural and, thus, relatively easily displaced1. In such cases, plaque healing and growth will ensue, and the thrombotic episode will pass either silently or with transient symptoms. Whether the attack is diagnosed as an episode of UA or NQMI will depend upon the duration and extent of occlusion caused by the thrombus: occlusion that is either partial or intermittent gives rise to the symptoms of UA, whereas NQMI is the result of either a more persistent partial occlusion or total occlusion in combination with collateral blood flow2,3. Regardless of the precise diagnosis, patients are at particularly high risk of recurrent events following an acute episode of UCAD4. The symptoms of UCAD arise from partial occlusion of the arterial lumen by the primary platelet-rich thrombus Unless adequately treated, patients with UCAD are at high risk of recurrence of acute events References 1 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441–460. 2 Spinler SA, Nawarskas JJ. Low-molecular-weight heparins for acute coronary syndromes. Ann Pharmacother 1998; 32: 103–110. 3 Turpie AGG. Low-molecular-weight heparins and unstable angina – current perspectives. Haemostasis 1997; 27 (suppl 1): 19–24. 4 Wallentin L, Husted S, Kontny F et al. Long-term low-molecular-weight heparin (Fragmin) and/or early revascularization during instability in coronary artery disease (the FRISC II Study). Am J Cardiol 1997; 80: 61E–63E. Adventitia
 Hyytymän muodostuminen. Thrombus. Lipid core. The formation of a platelet-rich, ‘white’ thrombus is the primary physiological response to rupture of an atherosclerotic plaque. If the initial disruption of the plaque is superficial, the thrombus formed will be mural and, thus, relatively easily displaced1. In such cases, plaque healing and growth will ensue, and the thrombotic episode will pass either silently or with transient symptoms. Whether the attack is diagnosed as an episode of UA or NQMI will depend upon the duration and extent of occlusion caused by the thrombus: occlusion that is either partial or intermittent gives rise to the symptoms of UA, whereas NQMI is the result of either a more persistent partial occlusion or total occlusion in combination with collateral blood flow2,3. Regardless of the precise diagnosis, patients are at particularly high risk of recurrent events following an acute episode of UCAD4. The symptoms of UCAD arise from partial occlusion of the arterial lumen by the primary platelet-rich thrombus. Unless adequately treated, patients with UCAD are at high risk of recurrence of acute events. References. 1 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441– Spinler SA, Nawarskas JJ. Low-molecular-weight heparins for acute coronary syndromes. Ann Pharmacother 1998; 32: 103– Turpie AGG. Low-molecular-weight heparins and unstable angina – current perspectives. Haemostasis 1997; 27 (suppl 1): 19–24. 4 Wallentin L, Husted S, Kontny F et al. Long-term low-molecular-weight heparin (Fragmin) and/or early revascularization during instability in coronary artery disease (the FRISC II Study). Am J Cardiol 1997; 80: 61E–63E. Adventitia.")
18
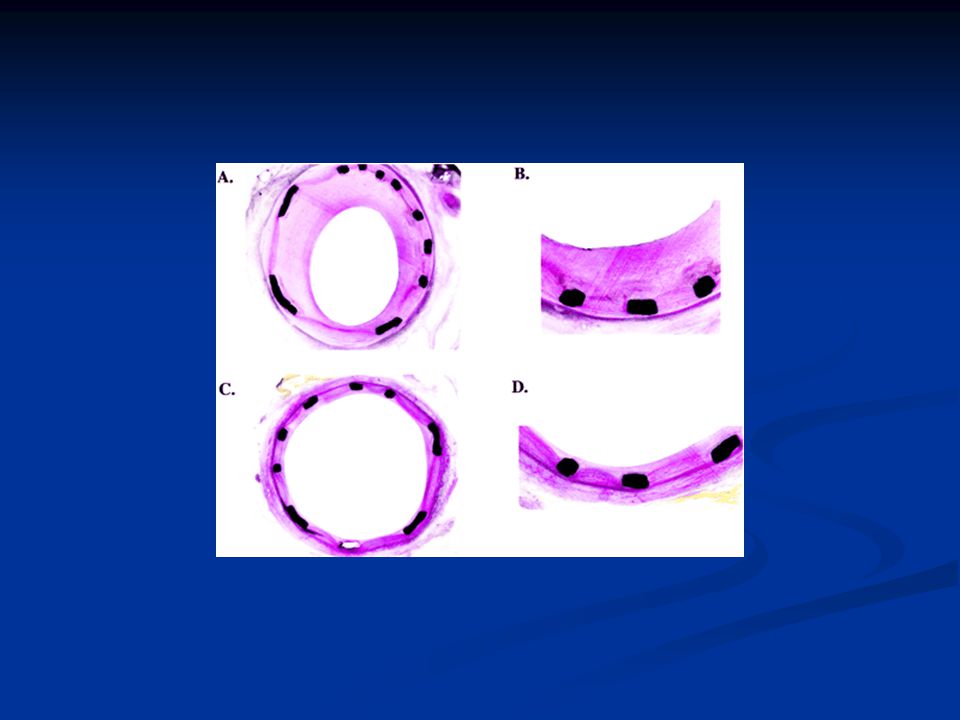
Revennyt plakki Pieni plakki Ohut, revennyt kansi ja trombi
Slide 23: The unstable atherosclerotic plaque Revennyt plakki Pieni plakki Ohut, revennyt kansi ja trombi Both images reproduced in this slide* depict an unstable atherosclerotic plaque, in which the fibrous cap has previously ruptured and is now overlaid with thrombotic material1,2. As discussed earlier, plaque rupture tends to occur at those regions where the infiltration of inflammatory cells3–6 and mechanical stresses on the fibrous cap are greatest – usually the shoulders of the plaque7,8. In the left hand image, dense macrophage infiltration is visible directly beneath the part of the cap that was subject to rupture and is now overlaid with thrombus. Note also the thinness of the fibrous cap – clearly apparent in both images. These images demonstrate that plaque rupture is not determined by size or degree of stenosis: the ruptured lesion depicted on the right is comparatively small and, with the cap intact, would barely extend into the arterial lumen. This is further underlined by the observation that 60–70% of all ACS events evolve from plaques that produce arterial stenosis of less than 50%9. Plaque rupture tends to occur at those points where inflammatory cell infiltration is highest and mechanical stresses are greatest – usually the shoulder regions of the plaque The likelihood of plaque rupture is not determined by size or degree of stenosis: 60–70% of all ACS events are caused by rupture of a plaque that produces less than 50% stenosis References 1 Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol 1997; 181 (1): 93–99. 2 Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998 (Figure 3-23): 81. 3 Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844–2850. 4 Shah PK. New insights into the pathogenesis and prevention of acute coronary syndromes. Am J Cardiol 1997; 79: 17–23. 5 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441–460. 6 Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657–671. 7 Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402–417. 8 Kristensen SD, Ravn HB, Falk E. Insights into the pathophysiology of unstable coronary artery disease. Am J Cardiol 1997; 80: 5E–9E. 9 Giri S, Waters DD. Pathophysiology and initial management of the acute coronary syndromes. Curr Opin Cardiol 1996; 11: 351–360. *Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol 1997; 181 (1): 93–99 Reproduced by permission of John Wiley & Sons Limited. *Image of ruptured lesion supplied by Professor Michael J Davies, British Heart Foundation Cardiovascular Pathology Unit, St George’s Hospital Medical School, London, UK Boyle et al, 1997 Runsaasti makrofaageja Davies and Ho, 1998
: 93–99. 2 Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998 (Figure 3-23): Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844– Shah PK. New insights into the pathogenesis and prevention of acute coronary syndromes. Am J Cardiol 1997; 79: 17–23. 5 Ross R, Fuster V. The pathogenesis of atherosclerosis. In: Fuster V, Ross R, Topol E. Atherosclerosis and Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1996: 441– Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657– Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res 1999; 41: 402– Kristensen SD, Ravn HB, Falk E. Insights into the pathophysiology of unstable coronary artery disease. Am J Cardiol 1997; 80: 5E–9E. 9 Giri S, Waters DD. Pathophysiology and initial management of the acute coronary syndromes. Curr Opin Cardiol 1996; 11: 351–360. *Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol 1997; 181 (1): 93–99 Reproduced by permission of John Wiley & Sons Limited. *Image of ruptured lesion supplied by Professor Michael J Davies, British Heart Foundation Cardiovascular Pathology Unit, St George’s Hospital Medical School, London, UK. Boyle et al, Runsaasti makrofaageja. Davies and Ho,")
19
Plakin kasvu Hajoava trombi Aiempi repeämä
Slide 24: Plaque growth Plakin kasvu Hajoava trombi Aiempi repeämä Uusien lihassolujen kertyminen Lipid core Plaque rupture is a common, but frequently clinically silent, complication in atherosclerosis1. Nevertheless, by way of its secretion of substances such as PDGF, basic fibroblast growth factor (BFGF) and transforming growth factor-ß (TGF-ß), the platelet-rich intraplaque thrombus formed when blood enters the lipid-rich core is a potent stimulus for VSMC proliferation and matrix synthesis2,3. Thus, any residual thrombotic material will invoke a new cycle of vigorous VSMC proliferation, collagen synthesis and plaque repair1,4. Such cycles of plaque rupture, thrombus formation, natural lysis and subsequent VSMC-driven repair may represent a major pathway for the progression of atherosclerotic lesions2,5. While this process ultimately restores stability to the disrupted plaque, it also increases the size of the lesion and may even lead to occlusion of the arterial lumen. Plaque rupture is a common, but frequently clinically silent, complication in atherosclerosis The platelet-rich thrombus formed in response to plaque rupture is a potent stimulus for VSMC proliferation, and any residual thrombotic material will invoke a new cycle of VSMC proliferation, collagen synthesis and plaque repair Such cycles of rupture and repair may represent a major pathway for atherosclerotic plaque growth References 1 Davies MJ, Ho SY. The acute ischemic syndromes and coronary disease progression. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 63–106. 2 Cimminiello C, Toschi V. Atherothrombosis: the role of platelets. Eur Heart J 1999; 1 (suppl A): A8–A13. 3 Prentice CRM. Platelets and atherosclerosis. Eur Heart J 1999; 1 (suppl A): A3–A7. 4 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 5 Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844–2850. Reprinted from Weissberg PL. Atherosclerosis involvement: more than just lipids – plaque dynamics. Eur Heart J 1999; 1 (suppl T): T13–T18, by permission of the publisher W.B. Saunders Company Adventitia Weissberg, 1999
 and transforming growth factor-ß (TGF-ß), the platelet-rich intraplaque thrombus formed when blood enters the lipid-rich core is a potent stimulus for VSMC proliferation and matrix synthesis2,3. Thus, any residual thrombotic material will invoke a new cycle of vigorous VSMC proliferation, collagen synthesis and plaque repair1,4. Such cycles of plaque rupture, thrombus formation, natural lysis and subsequent VSMC-driven repair may represent a major pathway for the progression of atherosclerotic lesions2,5. While this process ultimately restores stability to the disrupted plaque, it also increases the size of the lesion and may even lead to occlusion of the arterial lumen. Plaque rupture is a common, but frequently clinically silent, complication in atherosclerosis. The platelet-rich thrombus formed in response to plaque rupture is a potent stimulus for VSMC proliferation, and any residual thrombotic material will invoke a new cycle of VSMC proliferation, collagen synthesis and plaque repair. Such cycles of rupture and repair may represent a major pathway for atherosclerotic plaque growth. References. 1 Davies MJ, Ho SY. The acute ischemic syndromes and coronary disease progression. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 63– Cimminiello C, Toschi V. Atherothrombosis: the role of platelets. Eur Heart J 1999; 1 (suppl A): A8–A13. 3 Prentice CRM. Platelets and atherosclerosis. Eur Heart J 1999; 1 (suppl A): A3–A7. 4 Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10. 5 Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91: 2844–2850. Reprinted from Weissberg PL. Atherosclerosis involvement: more than just lipids – plaque dynamics. Eur Heart J 1999; 1 (suppl T): T13–T18, by permission of the publisher W.B. Saunders Company. Adventitia. Weissberg,")
20
Slide 25: The healed atherosclerotic plaque
Parantunut plakki As illustrated in this slide, repair of the ruptured cap of an atherosclerotic lesion can be effected with little or no additional encroachment into the arterial lumen1. The plaque shown here* still incorporates some residual thrombotic material, but a mass of new extracellular matrix material can be seen beginning to repair the rupture and replace the gap left by the lysed thrombus. Reference 1 Davies MJ, Ho SY. The acute ischemic syndromes and coronary disease progression. In: Davies MJ, Ho SY. Atlas of Coronary Artery Disease. Lippincott-Raven, Philadelphia, USA, 1998: 63–106. *Image supplied by Professor Michael J Davies, British Heart Foundation Cardiovascular Pathology Unit, St George’s Hospital Medical School, London, UK Davies and Ho, 1998

21
Ateroskleroosin tasapaino
Slide 40: The balance of atherosclerosis Lipidien lisääntyminen Lipidien hapettuminen Tulehdus? Perimä Lipidien vähentäminen Antioksidantit? PCI Repair Inflammation In conclusion, atherosclerosis is a dynamic process. Its progression is determined by the balance between factors and agents that promote inflammation and degradation, and those that encourage plaque stabilization and repair. While some of the factors involved in this dynamic equilibrium are now well established, others remain putative at this stage. What is clear, however, is that atherosclerosis is a multifactorial, dynamic disease, with different factors contributing to atherogenesis in different individuals. It must be hoped that, as further contributors to either side of the stability equation are identified, improvements in the available treatments will follow. However, implementation of the existing evidence is needed – that is, closure of the treatment gap in the secondary prevention of atherosclerotic disease by administering pravastatin. Atherosclerosis is a dynamic process, the progression of which is determined by the balance between inflammatory and stabilizing influences Atherosclerosis is a multifactorial disorder; hence different factors may contribute to the disease process in different individuals Reprinted from Weissberg P. Mechanisms modifying atherosclerotic disease – from lipids to vascular biology. Atherosclerosis 1999; 147 (suppl 1): S3–S10, with permission from Elsevier Science Stabiili plakki Epästabiili plakki
: S3–S10, with permission from Elsevier Science. Stabiili plakki. Epästabiili plakki.")
22
Sepelvaltimotaudin hoito
Ehkäisy Primaari- ja sekundaaripreventio Lääkehoito Anti-iskeeminen Riskitekijöihin kohdennettu Verenkierron palauttaminen ahtautuneen suonen alueelle eli revaskularisaatio Epästabiileissa tautimuodoissa kiireellisesti tai jopa päivystysluonteisesti Stabiileissa tautimuodoissa jos lääkehoidolla ei saada riittävää vastetta XI/99 KK

23
Epästabiili angina pectoris
I Äskettäin alkanut vaikea tai pahentunut angina, ei lepokipuja A) sekundaarinen B) primaari C) postinfarktiangina 2 viikon sisällä AMI:sta II Lepokipu < 1 kk mutta ei < 48 t (lepoangina, subakuutti) A, B, C kuten yllä III Lepokipu < 48 t (lepoangina, akuutti) troponiini – ja troponiini +, korreloi ennusteeseen Hamm & Braunwald; Circulation 2000;102:
 sekundaarinen. B) primaari. C) postinfarktiangina 2 viikon sisällä AMI:sta. II Lepokipu < 1 kk mutta ei < 48 t (lepoangina, subakuutti) A, B, C kuten yllä. III Lepokipu < 48 t (lepoangina, akuutti) troponiini – ja troponiini +, korreloi ennusteeseen. Hamm & Braunwald; Circulation 2000;102:")
24
Sydäninfarkti ilman ST-nousuja
NSTEAMI (Non ST-Elevation AMI) Iskeeminen kipu + merkkiainepäästö cTnT > 0.1 μg/l CK-MBm > 10 μg/l Ekg:ssa ST-lasku, nopeasti ohimenevä ST-nousu tai ei muutoksia yleensä ei kehity q-aaltoa Suomen Kardiologisen Seuran suositustyöryhmä. Duodecim 2000:116:
 Iskeeminen kipu + merkkiainepäästö. cTnT > 0.1 μg/l. CK-MBm > 10 μg/l. Ekg:ssa ST-lasku, nopeasti ohimenevä ST-nousu tai ei muutoksia. yleensä ei kehity q-aaltoa. Suomen Kardiologisen Seuran suositustyöryhmä. Duodecim 2000:116:")
25
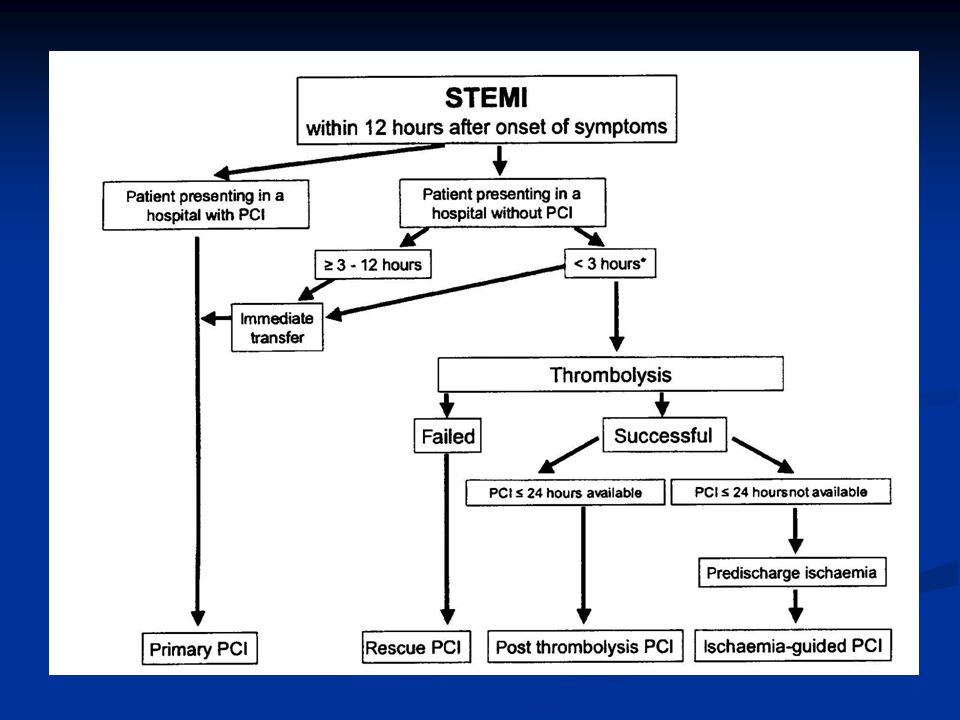
ST-nousuinfarkti STEAMI Täydellinen akuutti sepelvaltimotukos
Johtaa yleensä q-aaltoinfarktin kehittymiseen, jos tukos ei nopeasti avaudu

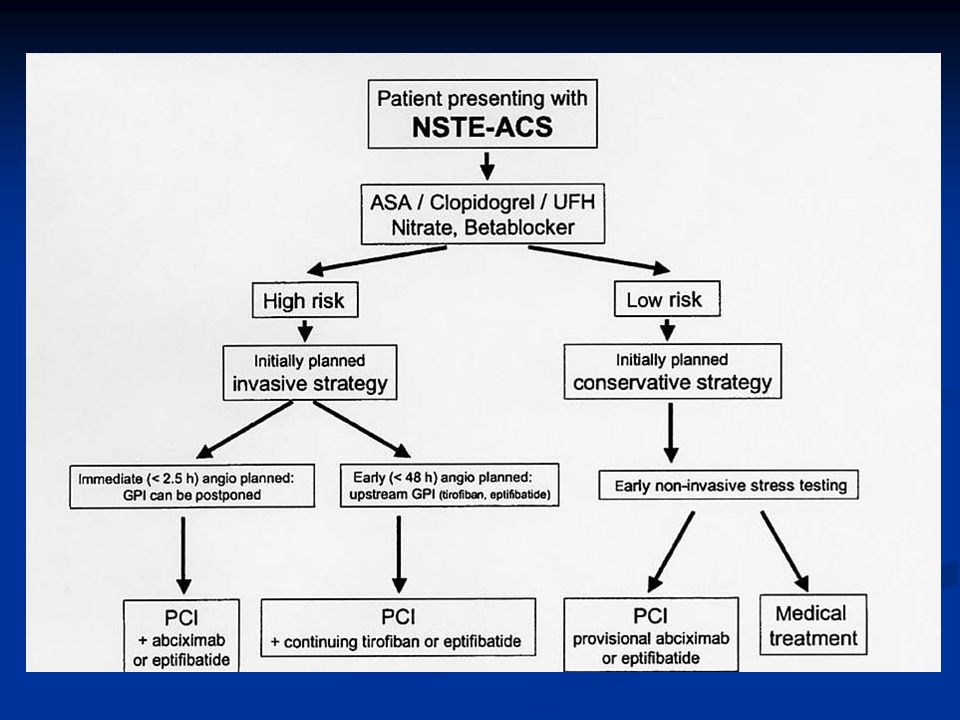
29
Varjoainekuvaus Varhainen suuren riskin potilailla, pyritään tekemään vrk:n kuluessa oireiden alusta kardiologin konsultaatio heti suuren riskin potilaista TaYS:n kardiologista varallaoloa käytetään tarvittaessa Pienen riskin potilailla lyhyen aikavälin ennuste hyvä, pitkän aikavälin ennuste arvioitava erikseen N. 20%:lla sepelvaltimotautikohtauksen vuoksi sairaalaan joutuneista angiografia normaali Kuvaus aiheellinen myös jos diagnoosi jää epävarmaksi

30
ST-nousuinfarktissa tavoitteena varhainen reperfuusio
Jos suoni auki 90 min => mortaliteetti 30 vrk 4.3% vs 9.1% (Gusto I) Onnistuu kemiallisella reperfuusiolla 50-60%:ssa mekaanisella reperfuusiolla 85-95%:ssa Kemiallinen reperfuusio pääsääntöisesti ensisijainen hoitolinja Viiveet minimoitava molemmissa hoitomuodoissa
 Onnistuu. kemiallisella reperfuusiolla 50-60%:ssa. mekaanisella reperfuusiolla 85-95%:ssa. Kemiallinen reperfuusio pääsääntöisesti ensisijainen hoitolinja. Viiveet minimoitava molemmissa hoitomuodoissa.")
32
PCI ST-nousu AMI:ssaVKS:ssa
Kun fibrinolyyttinen hoito on vasta-aiheinen Kun fibrinolyyttinen hoito epäonnistu (=rescue) suoni ei aukea 90 min kuluessa ekg:n ST-segmentin resoluutio < 70% (ST-nousujen summan väheneminen) hemodynaaminen tai sähköinen epästabilius Kardiogeeninen shokki sairaalaan tullessa Virka-aikana mahdollista aina Muulloin soitettava VKS:n sydänaseman kardiologille Kännykkänumerot löytyvät CCU:sta
 suoni ei aukea 90 min kuluessa. ekg:n ST-segmentin resoluutio < 70% (ST-nousujen summan väheneminen) hemodynaaminen tai sähköinen epästabilius. Kardiogeeninen shokki sairaalaan tullessa. Virka-aikana mahdollista aina. Muulloin soitettava VKS:n sydänaseman kardiologille. Kännykkänumerot löytyvät CCU:sta.")
33
PCI ST-nousu AMI:ssaVKS:ssa
Virka-aikana mahdollista myös: kaikki päivystyspkl:lle tulevat ST-nousuinfarktit, joille ei ole aloitettu fibrinolyysiä ensihoidon tai terveyskeskuksen toimesta ensihoidon ja terveyskeskusten lääkärit voivat konsultoida kardiologia siitä tehdäänkö primaari PCI vai annetaanko fibrinoolyysi Abciximabi tai eptifibatidi aloitetaan heti kun mahdollista

34
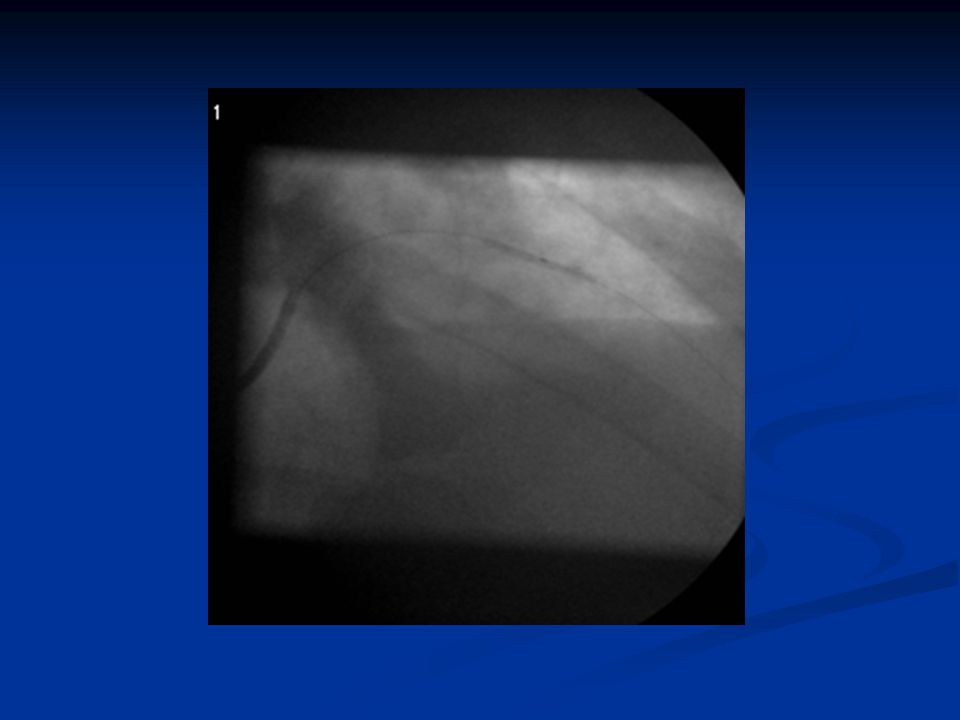
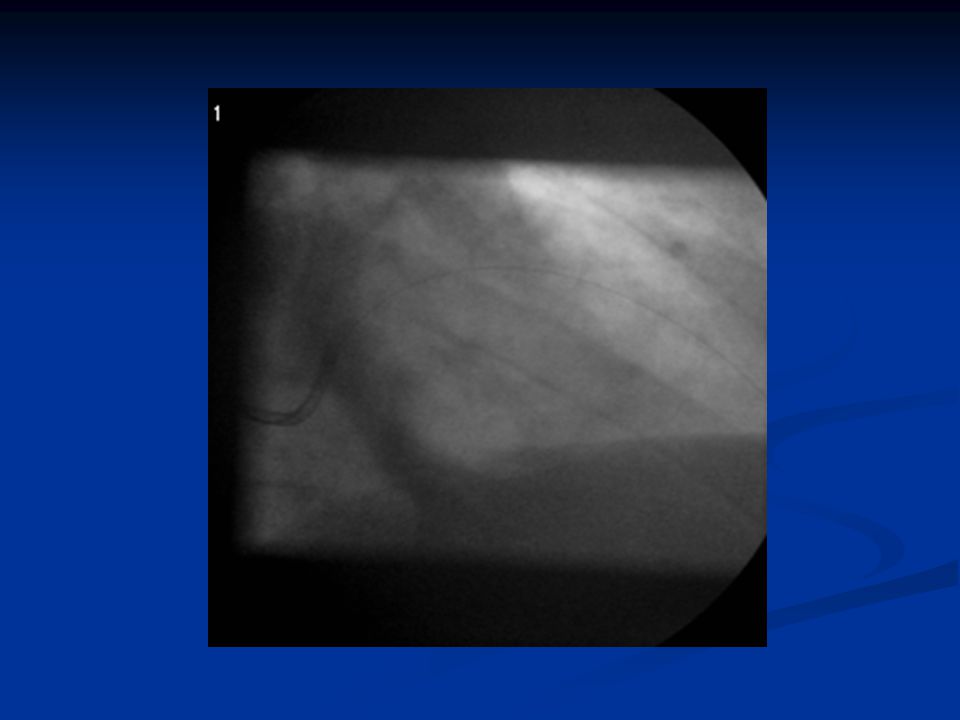
ST-nousuinfarktin angioplastia

38
Hoitoketju akuutin vaiheen jälkeen
Infarktipotilailla ensimmäinen kontrolli sisätautipk:lla kajoavien tutkimusten tarve (jos ei jo tehty) riskitekijöiden hoito kerrataan työkyky arvioidaan jatko avohoidossa Muut siirtyvät pääsääntöisesti suoraan avohoitoon
 riskitekijöiden hoito kerrataan. työkyky arvioidaan. jatko avohoidossa. Muut siirtyvät pääsääntöisesti suoraan avohoitoon.")
39
Sepelvaltimotautipotilaan lääkehoito
Oireenmukainen Ennusteellinen β-salpaaja antitromboottinen lääkitys lipidilääkitys ACE-estäjä Riskitekijöiden lääkehoito Lipidit, verenpaine, sokeritauti

40
β-salpaaja Suositus: pysyvästi kaikille sydäninfarktipotilaille, joilla ei kontraindikaatioita Tutkimusnäyttö ennustevaikutuksesta propranololilla, metoprololilla, timololilla, asebutololilla ja karvedilolilla Vain pindololilla kielteinen tulos

41
Antitromboottinen lääkitys
ASA 100 mg päivässä pysyvästi jos ei allergiaa Klopidogreeli kaikille ASA-allergisille pysyvästi kaikille kajoavasti hoidetuille 9-12 kk riskipotilaille pidempään? Kaikille joilla sydäninfarkti ilman ST-nousua 12 kk kaikille ST-nousuinfarktipotilaille ? Varfariini + ASA laaja akinesia, FA, kammiotrombi mahdollisesti hyötyvät

42
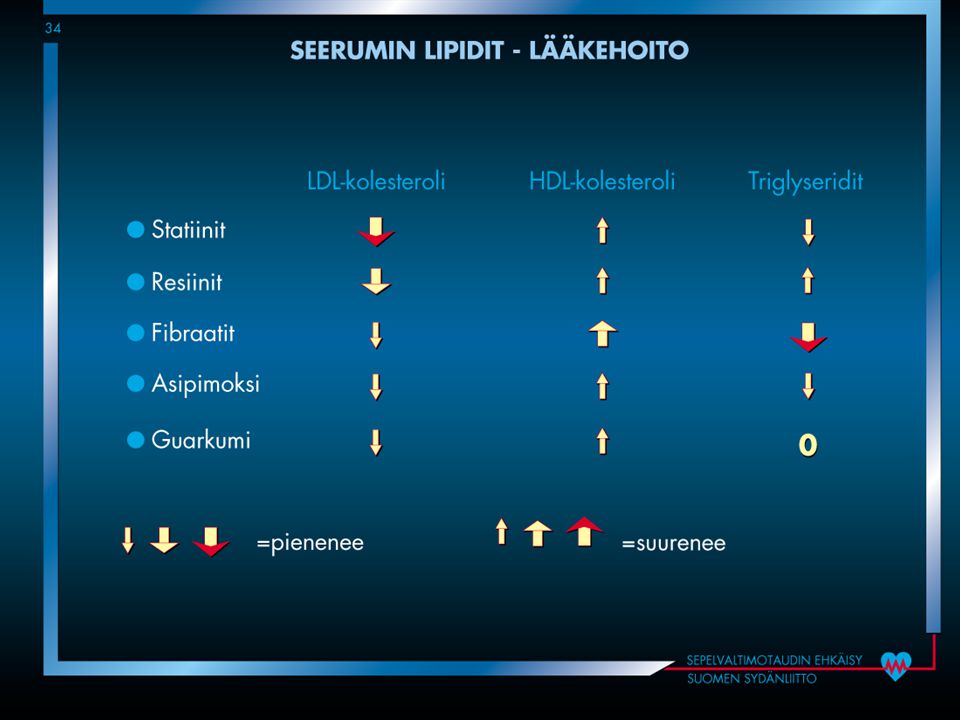
Lipidilääkkeet Hyperkolesterolemian hoitotavoiteet
Jos todettu valtimotauti Diabetes Korkea sairastumisriski (>5%/10 vuotta) kokonaiskolesteroli < 4.5 mmol/l LDL-kolesteroli < 2.5 mmol/l
 kokonaiskolesteroli < 4.5 mmol/l. LDL-kolesteroli < 2.5 mmol/l.")
43
Kolesterolitason kehitys

44
ACE-estäjät Ainakin jos Ehkä kaikille?
alentunut vasemman kamion systoolinen funktio ohimennyt vajaatoiminta akuuttivaiheessa diabeetikot hyötyvät ilmeisesti enemmän hyöty kestää ainakin 5 vuotta infarktin jälkeen pysyvästi jos hyvin siedetty? Ehkä kaikille? EUROPA-tutkimus: perindopriili vähensi sydäntapahtumia 20%:lla (10% vs 8%) matalan riskin sepelvaltimopotilailla
 matalan riskin sepelvaltimopotilailla.")
45
Stabiilioireisen sepelvaltimotaudin hoito
Anti-iskeeminen lääkehoito nitraatit beetasalpaajat kalsiumsalpaajat Revaskularisaatio katetritekniikat (”pallolaajennus”) ohitusleikkaukset XI/99 KK
 ohitusleikkaukset. XI/99. KK.")
46
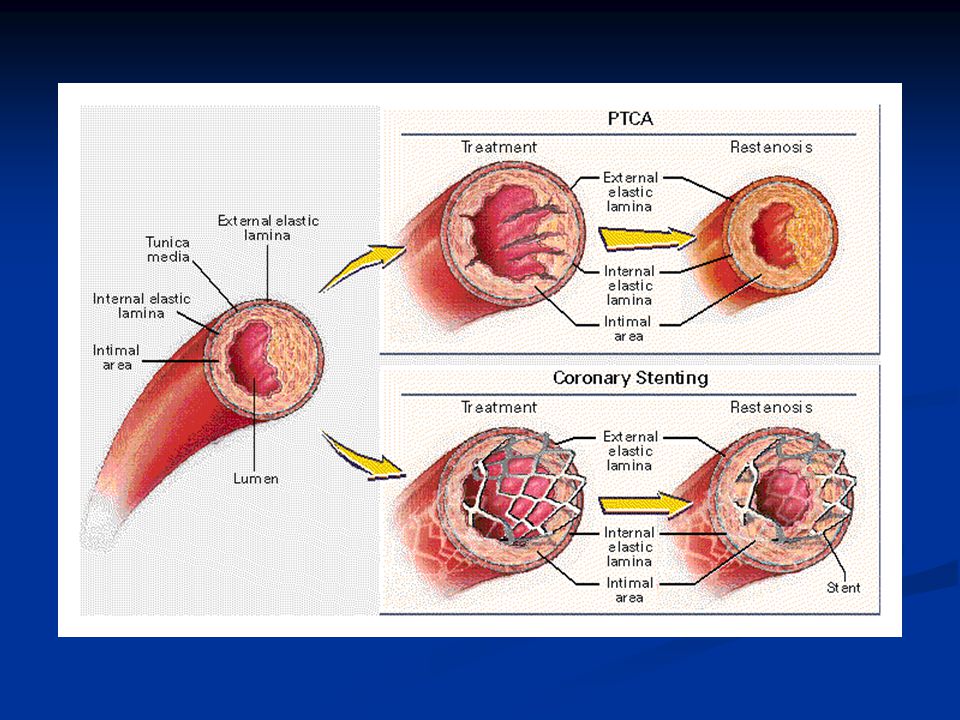
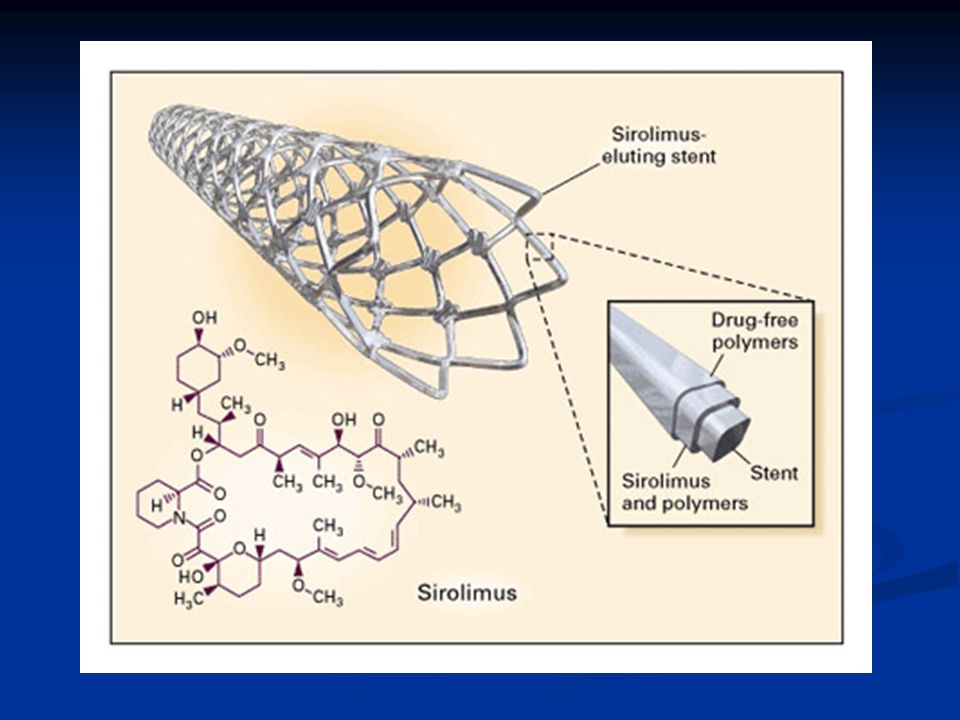
Sepelvaltimotaudin kajoavat hoitomahdollisuudet
Katetritekniikat Pallolaajennus Stentti Lääkeainepinnoitettu stentti Timanttiporaus Paikallinen sädehoito XI/99 KK

47
Sepelvaltimotaudin kajoavat hoitomahdollisuudet
Kirurgiset hoitomuodot ohitusleikkaus (CABG) leikkaus ”käyvällä sydämellä” mini-invasiivinen ohitusleikkaus sydämensiirto Yhdistelmähoidot mini-invasiivinen leikkaus + PTCA XI/99 KK
 leikkaus käyvällä sydämellä mini-invasiivinen ohitusleikkaus. sydämensiirto. Yhdistelmähoidot. mini-invasiivinen leikkaus + PTCA. XI/99. KK.")
48
Revaskularisaation aiheet
Ennusteen parantaminen vasemman sepelvaltimon päärungon ahtauma kolmen suonen tauti kahden suonen tauti kun mukana vasemman etulaskevan haaran tyviahtauma alentunut vasemman kammion toiminta + kahden tai kolmen suonen tauti XI/99 KK

50
Revaskularisaation aiheet
Oireiden helpottaminen asianmukaisesta lääkityksestä huolimatta kireä angina pectoris (NYHA/CCS toimintaluokka III tai IV) I – oireita vain ankarassa ruumiillisessa rasituksessa II – lievästi rajoittunut suorituskyky yli kahden korttelinvälin kävely tasamaalla enemmän kuin yhden kerrosvälin nousu III – huomattavasti rajoittunut suorituskyky IV - oireita kaikessa rasituksessa ja mahdollisesti myös levossa vähintään yksi merkittävä sepelvaltimoahtauma XI/99 KK
 I – oireita vain ankarassa ruumiillisessa rasituksessa. II – lievästi rajoittunut suorituskyky. yli kahden korttelinvälin kävely tasamaalla. enemmän kuin yhden kerrosvälin nousu. III – huomattavasti rajoittunut suorituskyky. IV - oireita kaikessa rasituksessa ja mahdollisesti myös levossa. vähintään yksi merkittävä sepelvaltimoahtauma. XI/99. KK.")
51
Leikkaus vai laajennus?
Laajennuksen etuja potilaan nopea toipuminen normaali verenkierto toimenpiteen aikana akuuteissa tiloissa nopea revaskularisaatio helppo uusia tarvittaessa Leikkauksen etuja varmemmin täydellinen tulos tietyissä tilanteissa uusintatoimenpiteiden tarve harvinaisempaa XI/99 KK

52
Leikkaus vai laajennus?
Laajennuksen ongelmia uudelleen ahtautuminen => uusintatoimenpiteiden tarve ei sovellu kaikille päärunkoahtauma suonten tyviosien ahtaumat vanhat täystukokset huono vasemman kammion toiminta XI/99 KK

56
Ohitusleikkaus Sydänlihaksen verenkierron parantaminen luomalla uusia väyliä verenkierrolle ohitesuonten avulla Laskimo-ohitteet aortasta sepelvaltimoihin Valtimo-ohitteet XI/99 KK

57
Anatomy of the Internal Thoracic Artery

59
Leikkaus vai laajennus?
Leikkauksen ongelmia keinotekoinen verenkierto leikkauksen aikana aivoverenkierto munuaisverenkierto suolistoverenkierto toipuminen kestää 3-6 kk uusintatoimenpiteissä lisääntynyt riski arpimuodostus toimivat valtimosiirteet XI/99 KK

60
Ohitusleikkaus Perinteisessä leikkauksessa sydän pysäytetään liitossaumojen teon ajaksi Sydänlihaksen suojaus (kardioplegia) Keinoverenkierto (sydän-keuhkokone) kudosten verenvirtaus ei vastaa fysiologista tilaa Mini-invasiivinen kirurgia Kirurgia käyvällä sydämellä XI/99 KK
 kudosten verenvirtaus ei vastaa fysiologista tilaa. Mini-invasiivinen kirurgia. Kirurgia käyvällä sydämellä. XI/99. KK.")
61
Jälkihoidossa keskeistä
Pitkällä aikavälillä ohitusleikkaus ohitesuonten tukkeutumisen ehkäisy sepelvaltimotaudin etenemisen ehkäisy: riskitekijät angioplastia

62
Ohitesuonten tukkeutuminen
Laskimo-ohitteet Valtimo-ohitteet 1.vuosi 15% 5% 5 vuotta 25% 5% 10 vuotta 50% %

63
Ehkäisy revaskularisaation jälkeen
Tupakoinnin lopettaminen Verenpaineen hoito Hyperlipidemian hoito kol < 4.5 LDL < 2.5 HDL > 1 Diabeteksen hoito Ylipainon välttäminen Liikuntaan kannustus

64
Tupakoinnin kehitys

65
Verenpaineen kehitys

66
Painoindeksin kehitys

68
Sepelvaltimotaudin sekundaaripreventio
Β-salpaaja Antitromboottinen lääkitys Hyperlipidemia Verenpainetauti Sokeritauti Ylipaino Liikunta

Samankaltaiset esitykset