Lataa esitys
Esittely latautuu. Ole hyvä ja odota

1
Proteiinianalyysi 2

2
folding problem Stability Specificity
How does the amino acid sequence encode the 3D structure? Any possible folding of the mainchain places different residues in contact. The interactions of the sidechains and mainchain, with one another and with the solvent, and the restrictions placed on sidechain mobility, determine the relative stabilities of different conformations. 2nd law of thermodynamics: Systems at constant temperature and pressure find an equilibrium state that is a compromise between comfort (low enthalpy, H) and freedom (high entropy, S), to give a minimum Gibbs free energy G=H-TS, in which T is the absolute temperature Specificity Proteins have evolved so that one folding pattern of the mainchain is thermodynamically significantly better than other conformations.
 and freedom (high entropy, S), to give a minimum Gibbs free energy G=H-TS, in which T is the absolute temperature. Specificity. Proteins have evolved so that one folding pattern of the mainchain is thermodynamically significantly better than other conformations.")
3
Some specific interactions:
Hydrogen-bonding: Hydrogen bound to Oxygen, Nitrogen and Sulfur has a smaller van der Waal's radius and more partial charge than hydrogen on carbon making the interaction between a charge or dipole with such "donar groups" stronger and more orientation dependent. Aromatic-Aromatic Interactions: The aromatic amino acid sidechains of phenylalanine, tyrosine and tryptophane prefer interactions with interplane angles of around 90 degrees. The interaction has a quadrupole-quadrupole character (distance dependence ~1/r5) with dependence on the interplane angle. Aromatic-Polar/Charged-Group Interactions: The charge separation of aromatics with ring hydrogens having partial positive charges and the pi electron system being partially negative, leads to interactions with O, S and N groups in proteins which are relatively strong (distance dependence ~1/r3 to 1/r4) and orientation dependent.
 with dependence on the interplane angle. Aromatic-Polar/Charged-Group Interactions: The charge separation of aromatics with ring hydrogens having partial positive charges and the pi electron system being partially negative, leads to interactions with O, S and N groups in proteins which are relatively strong (distance dependence ~1/r3 to 1/r4) and orientation dependent.")
4
Packing preferences Phe-Tyr

5
Hydrophobic Effect Entropic driving force for self-association of non-polar groups in an aqueous environment. Water cage around hydrophobic group Entropic gain on releasing water to the bulk solvent

6
stability and denaturation
proteins are marginally stable 20-60 kJ-mol, equivalent to one or two water-water hydrogen bonds unique native conformation == loss of entropy hydrophobic effect secondary structure (SS) formation compensates for water-protein hydrogen bonds of buried polar groups sidechain entropy > SS preferences compactness > van der Waals force
 formation compensates for water-protein hydrogen bonds of buried polar groups. sidechain entropy > SS preferences. compactness > van der Waals force.")
7
folding - summary all residues must be in stereochemically allowed conformations buried polar atoms must be hydrogen bonded to other buried polar atoms enough hydrophobic surface must be buried, and the interior must be sufficiently densely packed

8
Folding pathways initial formation of ”molten globule” containing some native secondary structure but without the tertiary structural interactions that lock the structure into its final conformation hierarchical condensation > low-energy pathway for structure assembly

9
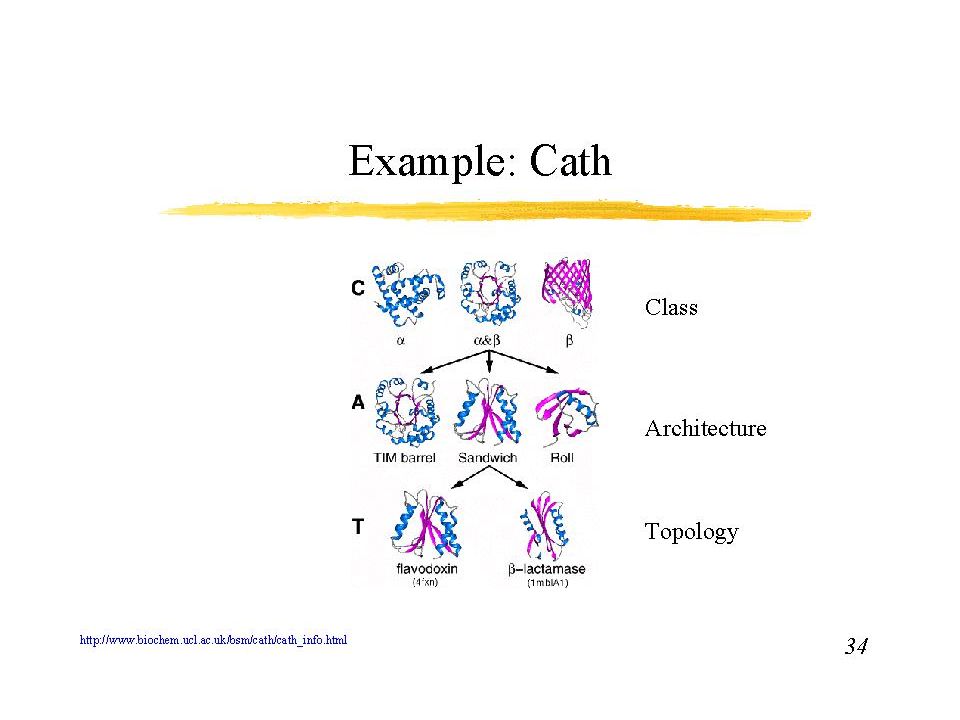
Topology diagrams There are a number of ways to represent the folding of a protein and the arrangement of secondary structure elements within the tertiary structure. While these simplifications don't show the sidechain and mainchain interactions that hold the structures together, they do reveal the folding pattern. Examination of such diagrams reveals recurring structural patterns in protein folding.

11
Arkkitehtuuri

12
Mainly alpha Up-down bundle Horseshoe Orthogonal bundle Solenoid

13
Efimov (1997) Proteins 28,
 Proteins 28,")
14
Mainly beta Ribbon Roll Single sheet Barrel

15
Clam Distorted sandwich 3-layer sandwich Sandwich

16
Prism Trefoil Aligned prism

17
6-propellor 5-propellor 7-propellor 4-propellor

18
8-propellor Complex 2-solenoid 3-solenoid

20
Mixed alpha-beta Roll Barrel Super roll Horseshoe

21
2-layer sandwich 3-layer bba sandwich 3-layer aba sandwich 4-layer sandwich

22
Alpha-beta prism Box Complex (Immunoglobulin/ Lipoprotein) 5-stranded propeller
 5-stranded propeller")
25
Complex (mixed alpha-beta) Irregular
 Irregular")
26
Yhteenveto - arkkitehtuuri
arkkitehtuuri = sekundaarirakenteiden yhteen pakkaaminen silmukoiden kiinnittyminen (topologia) jätetty huomiotta suositut arkkitehtuurit muistuttavat säännöllisiä geometrisia tiiveimpiä pakkauksia konvergentti evoluutio samanlaisiin laskoksiin Rakenne säilyy vaikka sekvenssit muuntelevat paljonkin – antaa evoluutiolle pelitilaa Hajautettu koodi: joistakin proteiineista on muutettu 30 % aminohapoista alaniiniksi laskoksen muuttumatta
 jätetty huomiotta. suositut arkkitehtuurit muistuttavat säännöllisiä geometrisia tiiveimpiä pakkauksia. konvergentti evoluutio samanlaisiin laskoksiin. Rakenne säilyy vaikka sekvenssit muuntelevat paljonkin – antaa evoluutiolle pelitilaa. Hajautettu koodi: joistakin proteiineista on muutettu 30 % aminohapoista alaniiniksi laskoksen muuttumatta.")
27
monen sekvenssin linjaus
evoluutiomallit

28
Multiple alignments provide more information than pairwise alignments
• Useful to confirm distant relationships • Provides a context for interpreting patterns of similarity and difference • "Speciation" over alignment space helps to connect and confirm widely degenerate motifs

33
Close relationships: Muscarinic receptors
Intermediate relationships: Prostaglandin receptors Distant Fungal pheromone receptors

37
Database searching • The first and most common operation in protein informatics...and the only way to access the information in large databases • Primary tool for inference of homologous structure and function • Improved algorithms to handle large databases quickly • Provides an estimate of statistical significance • Generates alignments • Definitions of similarity can be tuned using different scoring matrices and algorithm-specific parameters

38
Merkkijonojen vertailu
Hammingin etäisyys = mutaatioiden määrä, ei aukkoja Levenshteinin etäisyys = editointietäisyys = mutaatiot plus aukot substituutiomatriisi

39
Nyrkkisääntöjä >45 % identtisyys (koko domeenissa): lähes identtinen rakenne > 25 % identtisyys: samankaltainen rakenne Twilight zone (R.F.Doolittle): % identtisyys: homologia epävarmaa
: % identtisyys: homologia epävarmaa.")
40
Esimerkkejä myoglobiini / leghemoglobiini: 15 % identtisiä, homologisia rodaneesin N- ja C-terminaaliset domeenit: 11 % identtisiä, geeniduplikaatio kymotrypsiini / subtilisiini: 12 % identtisiä, samankaltainen aktiivinen keskus, konvergentti evoluutio

41
Log-odds scores Preferences for amino acid types at a given column
P = target distribution Q = background distribution S pi log pi / qi Assumes target distribution is at equilibrium Assumes column independence

42
q(i,j), i,j=1,…,20 observed frequency co-occurrence of amino acids
i,j at BLOCKS sites s(i,j)= 2 log2 (q(i,j)/e(i,j)) similarity score e(i,j)=2 p(i) p(j), if i≠j expected frequency of occurrence e(i,j)=p(i) p(i), if i=j p(i)=q(i,i)+S q(i,j)/2 frequency of occurrence of amino acid i≠j
= 2 log2 (q(i,j)/e(i,j)) similarity score. e(i,j)=2 p(i) p(j), if i≠j expected frequency of occurrence. e(i,j)=p(i) p(i), if i=j. p(i)=q(i,i)+S q(i,j)/2 frequency of occurrence of amino acid. i≠j.")
43
Example: calmodulin EF-loop
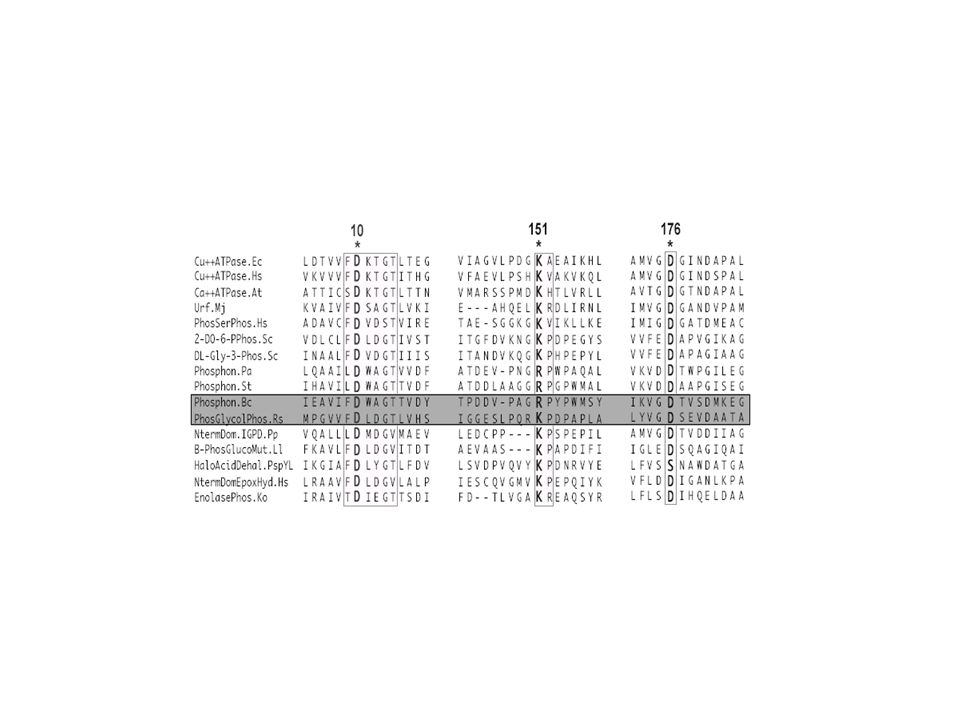
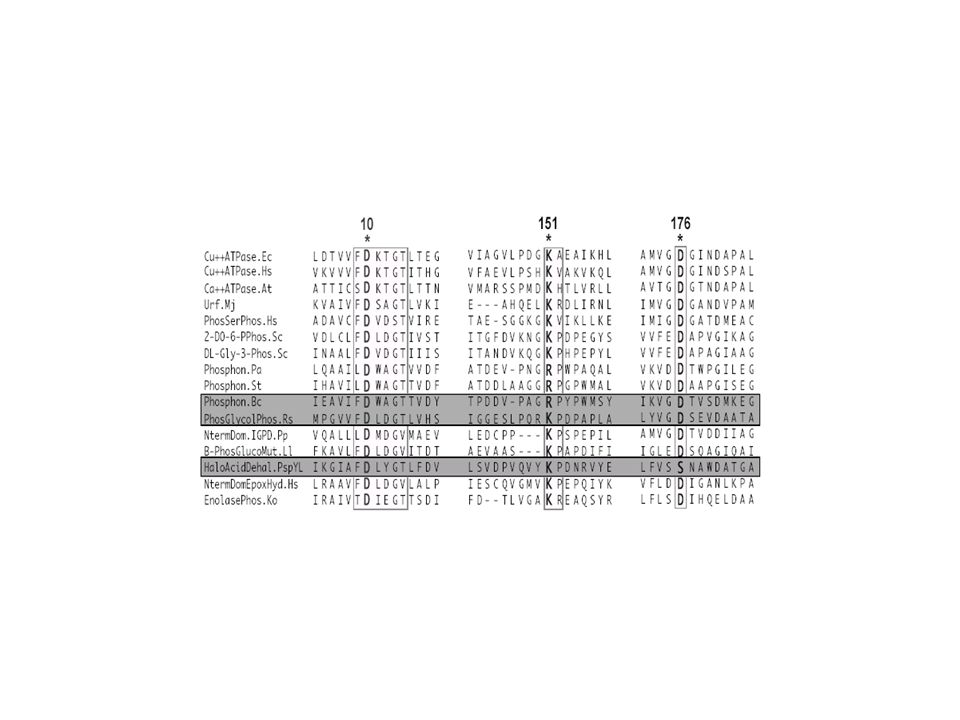
CALM_HUMAN_1 D K D G D G T I T T K E CALF_NAEGR_1 D K D G D G T I T T S E CALM_SCHPO_1 D R D Q D G N I T S N E CALM_HUMAN_2 D A D G N G T I D F P E CALF_NAEGR_2 D A D G N G T I D F T E CALM_SCHPO_2 D A D G N G T I D F T E CALM_HUMAN_3 D K D G N G Y I S A A E CALF_NAEGR_3 D K D G N G F I S A Q E CALM_SCHPO_3 D K D G N G Y I T V E E CALM_HUMAN_4 D I D G D G Q V N Y E E CALF_NAEGR_4 D I D G D N Q I N Y T E CALM_SCHPO_4 D T D G D G V I N Y E E 90 % Consensus D x D GD/NG x I x x x E

44
Sekvenssiprofiilit PSSM = position-specific scoring matrix
PSI-Blast, iteratiivinen HMM, piilomarkovmallit

45
Steps in a PSI-Blast search
• Constructs a multiple alignment from a Gapped Blast search and generates a profile from any significant local alignments found • The profile is compared to the protein database and PSI-BLAST estimates the statistical significance of the local alignments found, using "significant" hits to extend the profile for the next round • PSI-BLAST iterates step 2 an arbitrary number of times or until convergence

46
piilomarkovmalli (HMM)
probabilistinen malli aikasarjoille tai lineaarisille sekvensseille puheentunnistuksessa proteiiniperheiden mallitus kuvaa todennäköisyysjakaumaa sekvenssiavaruuden yli todennäköisyyksien summa = 1 generatiivinen malli sekvenssi voidaan linjata ja ”pisteyttää” mallia vastaan

47
Piilomarkovmalli kaksitilamuuttujalle
t=transitiotodennäköisyys p=emissiotodennäköisyys t(1,2) t(2,end) end 1 2 HMM p1(a) p1(b) p2(a) p2(b) tila, p 1 1 2 end a b a havaittu symbolisekvenssi, x t(1,1) t(1,2) t(2,end) p1(a) p1(b) p2(a) P(x,p | HMM)
 t(2,end) end HMM. p1(a) p1(b) p2(a) p2(b) tila, p. 1 1 2 end. a b a havaittu symbolisekvenssi, x. t(1,1) t(1,2) t(2,end) p1(a) p1(b) p2(a) P(x,p | HMM)")
48
profiili-HMM insert match delete begin 1 2 3 4 end
match state emits one of 20 amino acids insert state emits one of 20 amino acids delete, begin, end states are mute

49
profiili-HMM lineaarinen malli pisteytys vastaa log-odds-scorea
aukkosakkokin muodollisesti a+b(x-1) käyttö: monen sekvenssin linjaus parempi tulos saadaan esim. PSI-Blastilla homologien tunnistaminen: millä todennäköisyydellä HMM generoi testisekvenssin sekvenssin linjaus mallia vastaan
 käyttö: monen sekvenssin linjaus. parempi tulos saadaan esim. PSI-Blastilla. homologien tunnistaminen: millä todennäköisyydellä HMM generoi testisekvenssin. sekvenssin linjaus mallia vastaan.")
50
Types of alignment Sequence-sequence Sequence-profile Profile-profile
Target distribution = generic substitution matrix Sequence-profile Position-specific target distributions Profile-profile Observed frequencies from multiple alignment Average both ways Pair HMM Probability that two HMMs generate same sequence

Samankaltaiset esitykset















>")








